Two new schemes for computing molecular total atomization energies (TAEs)
and/or heats of formation (
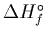
)
of first-and second-row
compounds to very high accuracy are presented. The more affordable
scheme, W1 (Weizmann-1) theory, yields a mean absolute
error of 0.30 kcal/mol and includes only a single, molecule-independent,
empirical parameter. It requires CCSD (coupled cluster with all
single and double substitutions) calculations in
spdf and
spdfg
basis sets, while CCSD(T) [i.e. CCSD with a quasiperturbative treatment
of connected triple excitations] calculations are only required in
spd and
spdf basis sets. On workstation computers and
using conventional coupled cluster algorithms, systems as large as
benzene can be treated, while larger systems are feasible using direct
coupled cluster methods. The more rigorous scheme, W2 (Weizmann-2) theory,
contains no empirical
parameters at all and yields a mean absolute error of 0.22 kcal/mol,
which is lowered to 0.17 kcal/mol for molecules dominated by dynamical
correlation. It involves CCSD calculations in
spdfg and
spdfgh basis
sets and CCSD(T) calculations in
spdf and
spdfg basis sets.
On workstation computers, molecules with up to three
heavy atoms can be treated using conventional coupled cluster algorithms,
while larger systems can still be treated using a direct CCSD code.
Both schemes include corrections for scalar relativistic effects,
which are found to be vital for accurate results on second-row
compounds.