


Next: Sample applications to larger
Up: W1 theory and its
Previous: Use of empirical extrapolation
We thus propose the following protocol for a
computational level which we will call W1 (Weizmann-1)
theory:
- geometry optimization at the B3LYP/VTZ+1 level
(B3LYP/VTZ if only first-row atoms are present). Alternatively,
the B3PW91 exchange-correlation functional may be preferable
for some systems like Cl2 -- under normal circumstances,
B3LYP/VTZ+1 and B3PW91/VTZ+1 should yield virtually identical
geometries;
- zero-point energy obtained from
B3LYP/VTZ+1 (or B3PW91/VTZ+1) harmonic frequencies scaled by 0.985;
- Carry out CCSD(T)/AVDZ+2d and CCSD(T)/AVTZ+2d1f
single-point calculations;
- Carry out a CCSD/AVQZ+2d1f single-point calculation;
- the SCF component of TAE is extrapolated by
A+B/Cl from SCF/AVDZ+2d, SCF/AVTZ+2d1f, and SCF/AVQZ+2d1f
components of TAE (l=2, 3, and 4, respectively)
- set
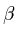 =3.22
=3.22
- the CCSD valence correlation component is obtained
from applying
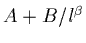 to CCSD/AVTZ+2d1f
and CCSD/AVQZ+2d1f valence correlation energies (l=3 and 4,
respectively).
In both
this and the next step, it is immaterial whether the extrapolation
is carried out on components to the total energy or to TAE;
to CCSD/AVTZ+2d1f
and CCSD/AVQZ+2d1f valence correlation energies (l=3 and 4,
respectively).
In both
this and the next step, it is immaterial whether the extrapolation
is carried out on components to the total energy or to TAE;
- the (T) valence correlation component is obtained
from applying
results to CCSD(T)/AVDZ+2d
and CCSD(T)/AVTZ+2d1f values for the (T) contribution.
- core correlation contributions are obtained at the
CCSD(T)/MTsmall level;
- scalar relativistic and, where necessary, spin-orbit
coupling effects are treated at the ACPF/MTsmall level. As in
W2 theory, this latter step can be combined in a single job
with the previous step.
W1 theory can be applied to fairly large systems (see below). CPU times
are dominated by the inner-shell correlation contribution (particularly
for second-row compounds), which is reflected in the relatively small
time reduction compared to W2 theory -- e.g., from 1h12' to 24' for
CO and from 13h42' to 8h48' for OCS. In addition -- contrary to
W2 theory -- W1 theory exhibits a pronounced difference in performance
between first-row and second-row compounds: for the species in Table VI,
MAE is 0.26 kcal/mol for first-row, but 0.40 kcal/mol for second-row
compounds. Since the CPU time gap between W1 and W2 theory is fairly
narrow for second-row species, we conclude that for accurate work on
second-row species -- unless precluded by disk space or memory
limitations -- it may well be worth to `walk the extra mile' and
carry out a W2 rather than a W1 calculation. For first-row systems,
on the contrary, W1 may well seem the more attractive of the two.
Next: Sample applications to larger
Up: W1 theory and its
Previous: Use of empirical extrapolation
Jan M.L. Martin
1999-04-20