PARE (Predicting Association Rate Enhancement)
The program PARE (Predicting Association Rate Enhancement) calculates the rate of association (kon) of mutant complexes from the Debye Huckel energy of interaction between proteins and an experimentally measured value of kon.
Theoretical background and experimental evidence for the power of PARE can be found in the following references:
- Selzer, T. and Schreiber, G. (1999). Predicting the Rate Enhancement of Protein Complex Formation from the Electrostatic Energy of Interaction. J. Mol. Biol. 287, 409-419. [Article (PDF)]
- Selzer, T., Albeck, S., and Schreiber, G.,(2000). Rational Design of Faster Associating and Tighter Binding Protein Complexes. Nat. Struct. Biol. 7, 537-541. [PubMed abstract]
- Selzer, T. and Schreiber, G. (2001). New Insights Into the Mechanism of Protein-Protein Association. Proteins.
Short background:
The rate of association is a function of an electrostatic independent
basal rate that is fixed for a given complex, and an electrostatic
dependent component that is a function of the Coulombic energy of
interaction -DU/RT (DU is the electrostatic energy of interaction. R is
the gas constant and T is the temperature). Combining these two gives:

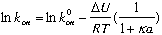
where a is set to 6A, and k is the Debye-huckel screening parameter that
relates to the ionic strength of the solution.
U is calculated using:
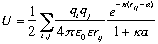
where i and j are the charged atoms in the proteins and e is the
dielectric constant.
The electrostatic energy of interaction is calculated
from the difference between the electrostatic energy of the complex and the electrostatic energy of the two individual proteins.
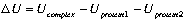
where Ucomplex and Uprotein are calculated from equation 2.
The relation between kon and DU at an ionic strength of 0.022M is:
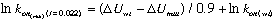
where lnkon(wt) has to be determined experimentally. (The adjustment of the rate to the rate at I=0.022 is done automatically by the program).
The energy calculation is performed using the following parameters:
 A constant dielectric constatnt set
to 80
A constant dielectric constatnt set
to 80
- The ionic strength of the solution is set
to 0.02M
- The program assigns charges only to atoms
of charged residues (ARG +1, LYS +1, ASP -1, GLU -1, C termnus (OXT) -1, N
terminus +1) Each full charge is distributed over one or two of the
atoms in the residue.

The association rate of a given charge mutated complex can be calculated in two ways:
- The accurate calculation: In which, the different charge mutations are already implemented in the input coordinate files of both reference protein complex and target protein complex. In this case the two input files will be different from each other and the input sets of charge rules will be the same. Most important: Although this method of calculation is more accurate then the following, special attention should be paid to the fact that when introducing charge mutations inside the binding site or in close vicinity to it, big differences in the calculated kon may appear for different conformers of the same charge mutation. Therefore it is recomended to perform several calculations for different conformers.[example]
- The coarse calculation: In which the charge mutations where not implemented in the input coordinates files of the reference protein complex, the target protein complex or both. In this case the input coordinate files of both reference and target proteins complexes are the same, and the differences between them will appear in giving two different sets of charge rules for each protein complex, in the charge input boxes.[example]
Note that introducing the correct charges is crucial for the calculation,
Therefore:
 The user must be sure that all side chain
atoms are present and if not, they should be added using any program that
is suitable for this task.
The user must be sure that all side chain
atoms are present and if not, they should be added using any program that
is suitable for this task.
- Each protein in the complex should be designated by a different chain letter.
- The C terminus atom of both proteins in the
protein complex should be given the name OXT (or else no charge will be
assigned to them).
- Substrates (ATP, GTP, GDP, SO42- etc...) and metals that appear in the coordinate files, their appropriate charge should be assigned.
- Always check the total charge of the protein
complex and individual proteins. If it does not fit your expectations,
check the coordinate file and charge input boxes again.
Developed by Tzvia Selzer in the group of Dr. Gideon Schreiber at the Dept. of Biological Chemistry at the Weizmann Institute of Science
 Back to the main window
Back to the main window